Metronidazol: Risiko schwerer Hepatotoxizität beim Cockayne-Syndrom
Last Updated on June 4, 2017 by Joseph Gut – thasso
03 Juni 2017 – Wie wir dem neuesten Newsletter der Pharmakovigilanz der Swissmedic, dem Schweizerischen Heilmittelinstitut entnehmen können, hält es Swissmedic für angebracht, betroffene medizinische Fachpersonen über das Risiko schwerer hepatotoxischer Reaktionen zu informieren, die bald nach Beginn einer systemischen Behandlung mit Metronidazol bei Patienten mit dem Cockayne-Syndrom auftraten.
Gemäss Swissmedic darf bei diesen Patienten Metronidazol nur nach einer sorgfältigen Evaluation des Nutzen-Risiko-Verhältnisses und nur dann angewendet werden, wenn keine alternative Behandlung zur Verfügung steht. Bei Patienten mit dem Cockayne-Syndrom kann schon kurz nach Beginn einer systemischen Verabreichung von Metronidazol zu Fällen schwerer Hepatotoxizität/akuter Leberinsuffizienz, auch mit tödlichem Verlauf, kommen. Daher sind bei Patienten mit dem Cockayne-Syndrom vor, während und nach der Behandlung mit Metronidazol Leberfunktionstests durchzuführen, bis die normalen Leberfunktionen wiederhergestellt oder die Zielwerte erreicht sind. Falls sich während der Behandlung deutlich erhöhte Leberwerte ergeben, muss die Behandlung abgebrochen werden. Patienten mit dem Cockayne-Syndrom sind darauf hinzuweisen, dass sie ihrem behandelnden Arzt jegliche Anzeichen einer beeinträchtigten Leberfunktion unverzüglich und zwingend melden müssen. Dazu gehören Bauchschmerzen, Anorexie, Übelkeit, Erbrechen, Fieber, Unwohlsein, Erschöpfung, Gelbsucht, dunkler Urin, entfärbter Stuhl oder Juckreiz. Die Metronidazol-Behandlung ist in diesem Fall abzubrechen.

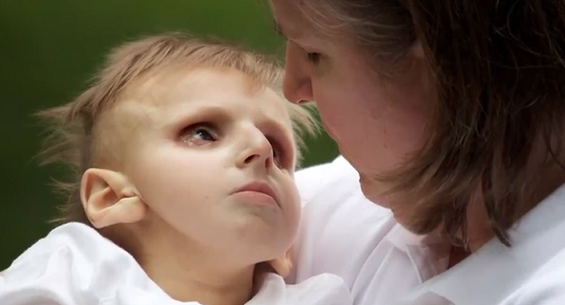
Das Cockayne-Syndrom (CS) ist eine seltene Krankheit, deren Prävalenz in Europa bei schätzungsweise einem Betroffenen pro 200’000 Geburten liegt. Es handelt sich um eine Erbkrankheit, deren Ursache für Cockayne B eine Mutation des ERCC6-Gens (auf Chromosom 10) und für Cockayne A eine Mutation des ERCC8-Gens (auf Chromosom 5) sind, die eine Anomalie des DNA-Reparatursystems und der DNA-Dekodierung (Transkription) zur Folge hat. Die Krankheit wird autosomal-rezessiv vererbt und kann sowohl bei Knaben als auch bei Mädchen auftreten. Das Cockayne-Syndrom zeigt sich klinisch durch Wachstumsstörungen, ein unterschiedlich ausgeprägtes intellektuelles Defizit, motorische Schwierigkeiten (neurologische Störungen) und eine Beeinträchtigung des Seh- und Hörvermögens. Die Kinder haben ein Gesicht, das vorzeitig gealtert scheint und sind auffallend mager (Kachexie). Die Symptome sind verschieden stark ausge- prägt und können in unterschiedlichem Alter auftreten.
Die häufigste Form des CS, die sogenannte klassische Form (Typ I), tritt während des ersten Lebensjahres mit Wachstumsverzögerung und neurologischen Störungen in Erscheinung, dann mit einer Verschlechterung des Seh- und Hörvermögens. Es gibt auch Fälle mit früherem Beginn und schwererem Verlauf (Typ II), wobei Augenanomalien und neurologische Störungen bereits ab Geburt auftreten. Schliesslich gibt es auch Fälle mit später auftretenden und weniger stark ausgeprägten Symptomen (Typ III). Das COFS-Syndrom (Zerebro-okulo-fazio-skelettales Syndrom) gehört zur bereits pränatal auftretenden klinischen Extremform des CS und ist durch sehr starke Missbildungen des Gehirns (Mikrozephalie), der Augen (Mikrophthalmie und Katarakt) und der Gelenke (Arthrogryposis multiplex congenita) gekennzeichnet. Bei einer weiteren Form des Cockayne-Syndroms treten alle Symptome des Syndroms zusammen mit Xeroderma pigmentosum und einer extremen Empfindlichkeit der Haut und der Augen gegenüber ultravioletter Strahlung (UV) auf, die mit starken Hautläsionen und einem erhöhten Hautkrebsrisiko verbunden sind. Es gibt derzeit keine Behandlung, mit der die Krankheit geheilt werden kann. Die Betreu- ung der Patienten beschränkt sich auf die Be- handlung der Symptome.
Related articles across the web






Related posts
Schwere Missbildungen bei Kindern nach Einnahme von Valproat (Depakine) in der S...
Was sagen Gene über schwarzen Kaffee und dunkle Schokolade aus?
Abartig: Wieso werden schwangere Frauen mit Sildenafil (Viagra) behandelt?
APOE Missense-Variante mit erhöhtem Alzheimer-Risiko bei Afro-Amerikanern
Genotyp-Zuerst: Voraussage genetisch bedingter Erkrankungen. Ein neuer Ansatz?
Professor in Pharmakologie und Toxikologie. Experte in theragenomischer und personalisierter Medizin und individualisierter Arzneimittelsicherheit. Experte in Pharmako- und Toxiko-Genetik. Experte in der klinischen Sicherheit von Arzneimitteln, Chemikalien, Umweltschadstoffen und Nahrungsinhaltsstoffen.